
Fra 31. januar 2022 vil klinisk utprøving/kliniske studier reguleres under et nytt europeisk regelverk, Forordning (EU) Nr. 536/2014, med mål om å blant annet harmonisere saksbehandlingen av klinisk utprøving innen Europa, øke sikkerheten til personer som deltar i klinisk utprøving, og sikre åpenhet rundt data fra klinisk utprøving. Det vil innebære en rekke administrative endringer for både søker og myndigheter, skriver Legemiddelverket.
Se oversikt over diverse forkortelser nederst i saken
Blant annet innebærer det innføringen av et felles digitalt saksbehandlingssystem kalt the Clinical Trials Information System (CTIS).
Ny ordning – mange spørsmål
Naturlig nok har legemiddelindustrien mange spørsmål om hvordan ting skal løses, rent praktisk. LMIs ClinOps-gruppe sendte derfor en rekke spørsmål til Legemiddelverket og REK, og i et Teams-møtetidligere i september ble spørsmålene gjennomgått. Over 40 personer deltok i møtet, som ble ledet av Margrete Vidringstad Bjurstrøm fra Boehringer Ingelheim/LMIs ClinOps-gruppe og Ingvild Aaløkken fra Legemiddelverket. Aaløkken hadde med seg kolleger fra teamet som jobber med legemiddelstudier og medisinsk utstyr. Fra REK stilte representanter fra komitéen for klinisk utprøving av legemidler, REK-KULMU.
Legemiddelverket startet møtet med å bekrefte at forordningen trer i kraft den 31. januar 2022, og at de har deltatt på opplæring i det nye systemet (Clinical Trials Information System (CTIS)). REK og SLV har dessuten fått god erfaring gjennom VHP og VHP Plus (Voluntary Harmonisation Procedure), et pilotprosjekt som har hatt som mål å forberede både myndighetene og industrien på forordningen. VHP og VHP Plus er prosedyre der man kan få en samordnet vitenskapelig vurdering av en klinisk legemiddelutprøving som skal gjennomføres i flere land i EU/EØS. VHP pluss innebærer at etikk-komiteene også deltar i vurderingen. Når forordningen trer i kraft, avsluttes denne pilotordningen.
– Vi må avslutte VHP og VHP Pluss 15. oktober for å rekke å bli ferdige med saksbehandlingen i dette systemet før forordningen trer i kraft, sa Aaløkken.
Hun fortalte også at EU-kommisjonen jobber med en såkalt implementing regulation for felles sikkerhetsvurderinger i Europa, som trolig kommer på høring i høst. Norge har representanter i CTEG (Clinical Trial Expert Group), både fra Legemiddelverket og REK-KULMU.
– Følg gjerne med på kommisjonens eget spørsmål og svar-dokument, det oppdateres fortløpende, sier Aaløkken.
Les også mer på EMAs nettsider
Forordningen innføres med en overgangsordning på tre år.
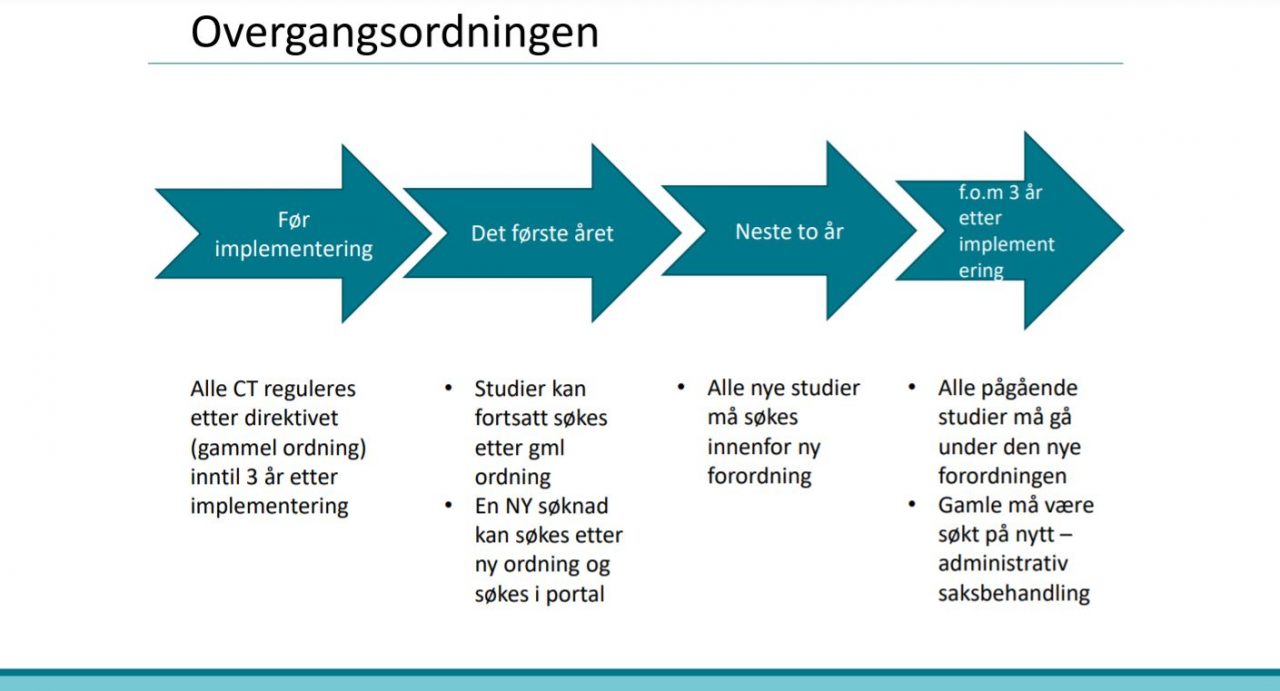
Under følger spørsmålene fra industrien og svarene fra myndighetene:
Hva er erfaringene fra piloten (VHP) som har blitt gjennomført?
SLV: Vi har gjennom VHP samarbeidet med andre medlemsstater i EU. Dette er en erfaring som vil komme oss til nytte. Vi som medlemsstater har ulik innfallsvinkel til søknadene og det gjenspeiles ofte i spørsmålene. Erfaringen tilsier også at vi har ulik forståelse for fortolkningen av regelverket og dokumenter. Men har nå fått jobbe med harmonisering av disse tingene. Vi har vært RMS (Reference Member State/referanseland) for noen prosedyrer, og der har vi lært at det innebærer en stor koordineringsjobb, særlig der det er mange medlemsstater involvert. Det har utfordret oss både administrativt og regulatorisk. Vi har samarbeidet med etikkomiteen her i Norge, og det har vært en nyttig og lærerik erfaring vi tar med oss videre.
REK KULMU: Vi deltok i VHP pluss fra høsten 2020 og har på denne relativt korte tiden fått nyttig erfaring både innad i REK og i samhandlingen med SLV. Vi føler oss derfor godt forberedt til CTR som trer i kraft neste år.
Når vil SLV og REK være «klare» til å ta imot søknader i CTR?
SLV: Vi er alle klare fra 31. januar 2022, når forordningen trer i kraft.
Vil alle norske dokumenter (både til SLV og REK) lastes opp i CTIS-portalen?
SLV: Ja, alle dokumenter som er relevant og kreves av fordringen skal lastes opp der.
Skal alle dokumentene lastes opp samtidig? Eks pasientinformasjon på forskjellig språk?
SLV: Det som tilhører del I (part I) av søknaden skal lastes opp samtidig. Søker kan velge om del II (part II) skal søkes samtidig eller på et annet tidspunkt. Når del II sendes skal alle dokumenter sendes samtidig her også.
Hvordan vil kommunikasjonen mellom SLV og REK KULMU være?
SLV: Vi vil ha tilstrekkelig og fortløpende kommunikasjon for å løse sakene, enten via møter, på epost, i felles saksbehandlingssystem eller på telefon hvis det blir nødvendig.
Når vil den nye REK KULMU-komiteen for legemidler være på plass og funksjonell?
REK: REK KULMU består nå av to komitéer; A og B. REK KULMU A ble opprettet i fjor høst, og har siden da jobbet med VHP Pluss og behandling av søknader vedrørende medisinsk utstyr som reguleres av MDR-forordningen (Forordning (EU) nr. 2017/745 om medisinsk utstyr) som trådte i kraft 26. mai i år. REK KULMU B har nylig blitt opprettet og vi skal prøve å få satt den i gang nå snart, antar innen et par måneders tid. Begge komitéene vil jobbe med å behandle legemiddelutprøvinger og utprøvinger om medisinsk utstyr.
Vi dere samkjøre mellom de to komiteene? Vi kan oppleve store forskjeller i tilbakemeldinger fra de ulike REK-komiteene, og hadde satt pris på en mer enhetlig kommunikasjon
REK: Både REK KULMU A OG B har samme lagleder, som vil være med på samtlige komitémøter fremover for å se til at like spørsmål besvares mest mulig likt. Vi skal prøve å få til en enhetlig praksis. Samtidig skal komiteene foreta en konkret helhetsvurdering av en studie. Ofte kan to studier se tilsvarende like ut, men etter en nærmere vurdering fremstå som ulike likevel. Men utgangspunktet er at like saker behandles likt, så vi har fokus på det.
Hva har skjedd med REK MIDT, REK SØR, REK ØST og REK VEST, er det slått sammen?
REK: REK KULMU A og B har blitt opprettet for å behandle studier under MDR (Medical Device Regulation), CTR (Clinical Trial Regulation) og IVDR (In-vitro Diagnostic Medical Device Regulation). De er opprettet med et nasjonalt mandat, som er lagt til REK SØR-ØST som har kontorer i Nydalen i Oslo. De ordinære komiteene består fortsatt, som vil behandle søknader som ikke faller inn under de nevnte forordningene.
Vil nasjonal koordinator for en ny studie allikevel måtte sende noe inn i REK-portalen, eller sendes hele REK-søknaden, inkludert pasientinformasjon, med hovedsøknaden?
REK: Søknadene skal innsendes til CTIS, både dokumenter i part I og part II. Forskningsprotokoll og IB skal innsendes i part I. Informasjons- og samtykkeskriv skal blant annet innsendes i part II.
Vil svar komme både fra REK og SLV samtidig?
REK og SLV: Ja, når det gjelder svar på søknad vedrørende part I. Svar på søknad i forbindelse med part II kan komme på et senere tidspunkt. Det beror på hvorvidt søknad til part I og part II sendes inn samtidig fra sponsor. Når konklusjonene for part I og part II er ferdigstilt, har hvert deltakerland inntil 5 dager til å sende beslutningen til sponsor. Beslutningen blir da et felles svar fra både REK og SLV.
Hvem bestemmer hvilke studier som skal søkes i henhold til CTR – det enkelte firma, eller myndighetene?
SLV: Det første året kan sponsor velge hvilket regelverk søknaden sendes inn etter.
Er det mulig å skissere hvordan CTIS-portalen praktisk skal fungere? Hvem sender hva hvor etc?
REK: Dere som søkere må være registrert med en EMA-brukerkonto, så det må dere få på plass før dere sender inn en søknad. Vi anbefaler alle å gå inn på EMAs nettsider og se på det omfattende opplæringsmateriellet som ligger ute. Både REK og SLV er med i en gruppe der vi får en del opplærling underveis om hvordan vi skal benytte systemet når det går live i januar. Mesteparten av saksbehandlingen vil skje i CTIS fra 2022, og der finnes det også et eget arbeidsområde for sponsorer. I CTIS er det mulig å sende inn søknader med tilhørende dokumentasjon, oppdatere dokumentene man sender inn, og her vil mesteparten av kommunikasjonen mellom myndighet og søker skje. Et referanseland får hovedansvaret for å utføre selve prosessen med å samle inn svar fra landene som er med, og også denne prosessen vil i hovedsak skje i CTIS.
Hva er forskjellen på Part 1 og Part 2-dokumenter?
REK: Part I-dokumenter er grunnlaget for nytte-risikovurderingen av studien, der er både SLV og REK med i vurderingsprosessen. Part 2-dokumenter det først og fremst REK som vil se på. Årsaken til at delt inn slik er at Part 2 gjelder mer spesifikt for hvert enkelt land, investigators cv, interessekonflikter, ulike siters egnethet for å kunne være med i studien, rekrutteringsprosedyre, pasientinformasjon, samtykkeskjema, informasjon om betaling for kompensasjon til deltakerne mm.
Går all kommunikasjon i systemet? Hva hvis vi har spørsmål?
SLV: Ja, all kommunikasjon skal skje i CTIS-systemet, og man får notifikasjoner der når det har vært en oppdatering eller man har fått spørsmål/henvendelser. Så ja, det skal skje i CTIS. Man må dessverre fysisk inn i systemet for å se notifikasjonene, man får ikke et e-postvarsel om dette. Det gjelder myndighetene også.
Ved en felles søknad hvor et eller flere av landene som vurderer søknaden mener produktet er ett GMO, mens andre land ikke er av den oppfatning, hva skjer da med godkjenningen?
SLV: Det vil måtte bli to separate søknader, og da må man søke GMO-myndighetene i det aktuelle landet. Man må ha godkjenning i det enkelte land.
Hvis avslag, blir da hele søknaden avslått?
SLV: Delegering av myndighet for miljørisikovurdering av GMO-legemidlet overføres fra Klima- og miljødepartementet til Helse- og omsorgsdepartementet. I praksis vil det si at delegering av myndighet flyttes fra Miljødirektoratet til Legemiddelverket. Endringene trer i kraft 15. november 2021.
Kongens myndighet etter lov 2. april 1993 nr. 38 om framstilling og bruk av genmodifiserte organismer m.m. (genteknologiloven) § 10 første ledd overføres fra Klima- og miljødepartementet til Helse- og omsorgsdepartementet for så vidt gjelder kompetanse til å godkjenne søknader om klinisk utprøving av GMO-legemidler.
Delegering av Kongens myndighet etter genteknologiloven § 10 første ledd til Klima- og miljødepartementet, gitt ved kongelig resolusjon 20. august 1993, oppheves for så vidt gjelder kompetanse til å behandle søknader om klinisk utprøving av GMO-legemidler.
Må du vente på svar i CTIS?
SLV: Svaret på miljørisikovurderingen av GMO-legemidlet vil komme fra den myndighet som har ansvaret for miljørisikovurderingen. Dette svaret kommer separat fra vurderingen av utprøvingen, og kommer ikke i CTIS, men i ordinær post/e-post.
Jobbes det med å implementere søknad om vurdering av miljørisiko for GMO-legemidler i CTR og innsending til CTIS?
SLV: Vi har ingen informasjon som tilsier at søknad om vurdering av GMO-legemidlet vil bli tatt inn forordningen for klinisk utprøving, og dermed inn CTIS. Foreløpig dreier diskusjonene seg om å få mer og bedre funksjonalitet i CTIS rettet mot vurdering av den kliniske utprøvingen.
Hva er erfaringene fra MTU-området så langt?
SLV: MTU er ikke et begrep vi bruker i SLV, i regelverket benyttes det kun «medisinsk utstyr». Vi tar imot søknader om medisinsk utstyr og saksbehandler de søknader som omfattes av MDR-forordningen som ble implementert 26. mai i år. Saksbehandlingstid kommer an på utstyrets klassifisering. Lavrisikoutstyr (klasse I og ikke-invasivt klasse IIa og IIb) skal kun valideres mens høyrisikoutstyr (invasivt klasse IIa/IIb/III) skal både valideres og vurderes. Validering utføres normalt innen 15 dager. Legemiddelverket og REK har 45 dagers saksbehandlingstid for vurderinger med mulighet til 20 dagers forlengelse ved bruk av eksterne eksperter. Identiske søknader sendes per dags dato inn både til REK, via REK-portalene, og på mail til SLV. Søknadene må sendes inn til REK og Legemiddelverket samme dag. Dette er en løsning vi benytter i påvente av at den nye EU-portalen (EUDAMED), som er forsinket, skal ferdigstilles. Søker skal på sikt sende én søknad inn via EUDAMED.
Hvordan vil evalueringen av studiesøknader fordeles mellom land?
SLV: Når søknaden sendes inn, er det sponsor som foreslår hvem de ønsker som ansvarlig utrederland.
Er det tatt noen beslutning fra Helse- og omsorgsdepartementet om hva studieavgiften blir dersom Norge er rapportørland, og dersom Norge er deltakerland? Og vil det påløpe studieavgift ved endringssøknader?
SLV: Det er ikke tatt beslutning enda, det blir trolig en høring etter hvert.
Bruk av templater utarbeidet av EU CTEG (Clinical Trial Expert Group): Vil alle templatene tillates brukt i Norge? Er det noen templater som ikke inneholder tilstrekkelig informasjon i henhold til krav fra norske myndigheter?
REK: Vi i REK KULMU vil i høst gjøre en grundig og systematisk gjennomgang av templatene. Vårt utgangspunkt er at vi finner det mest hensiktsmessig å benytte templatene. Vi kan eventuelt legge på tilleggskrav. Vi skal se grundig på dette i tiden som kommer. Vi kan også nevne at informasjonsskrivet i søknadens Del 2 må være på norsk. Vi vil trolig utarbeide oppdatert templat for informasjonsskriv som vil være i overensstemmelse med forordningen, og det vil legges ut oppdatert mal i REK-portalen.. Men vi skal altså ta et dypdykk i dette nå i høst.
Forskningsbiobank/ bruk av biologisk materiale: Vil norske myndigheter ha spesielle krav angående forskningsbiobanker? Hvordan ønsker norske myndigheter at bruk av biologisk materiale beskrives?
REK-KULMU: Med forbehold om at det ikke er bestemmelser i forordningen som direkte regulerer opprettelse av forskningsbiobanker i forbindelse med kliniske utprøvinger av legemidler, vil det etter REK KULMU sin oppfatning være helseforskningsloven som gjelder ved søknad om opprettelse av forskningsbiobanker i Norge. Bestemmelsene helseforskningsloven antas å være kjent for LMI, slik at allerede etablert praksis vil kunne videreføres ved søknader om forhåndsgodkjenning av kliniske utprøvninger av legemidler. Vi viser i denne sammenheng helseforskningsloven, Kapittel 6. Forskningsbiobanker og forskning som involverer humant biologisk materiale.
REK KULMU ønsker å videreføre den etablerte praksisen hvor innsamling, oppbevaring, behandling og bruk av humant biologisk materiale beskrives i forskningsprotokollen og i informasjonsskrivet som gis ut til forskningsdeltakerne. Informasjonen som gis til forskningsdeltakerne må nødvendigvis forenkles med tanke på bruk av medisinske ord og uttrykk, men må gjenspeile den planlagte behandlingen av materiale som er beskrevet i forskningsprotokollen.
REK har i sin saksbehandling av søknader om forhåndsgodkjenning av legemiddelutprøvinger erfart at det ofte søkes om godkjenning til utsendelse eller overføring av innsamlet humant biologisk materiale til biobanker etablert i andre land. Vilkårene for utsendelse følger av helseforskningsloven § 29. Er det planlagt en overføring av humant biologisk materiale til biobanker i utlandet, må dette komme tydelig frem i informasjonsskrivet som gis til forskningsdeltakerne. I prinsippet vil de samme vurderingene som institusjonen er pålagt å gjøre ved utsendelse av personopplysninger også gjøre seg gjeldende ved utførsel av humant biologisk materiale. Dette innebærer at forskningsansvarlig institusjon må sørge for å ha et rettslig grunnlag både for oppbevaring, bruk og behandling av materialet, inkludert et overføringsgrunnlag.
Møter hvor representanter fra industrien og studiesentre kan delta: Er det fastsatt møtetidspunkt for informasjon om søknadsprosessene/ lokale krav til industri/ studiesentre?
SLV: Det er vi i planleggingsfasen for nå, og vi planlegger å få til noe sent på høsten. Vi legger ut informasjon om dette på nettsiden vår når en dato er klar.
Forkortelser
REK: Regional etisk komite
REK KULMU: Regional komité for medisinsk og helsefaglig forskningsetikk ved komiteene for Klinisk utprøving av legemidler og medisinsk utstyr.
CTR: Clinical Trial Regulation
CTIS: Clinical Trials Information System
VHP: Voluntary Harmonisation Procedure
VHP Pluss: Voluntary Harmonisation Procedure der etikk-komiteene også deltar i vurderingen
CTEG: Clinical Trial Expert Group
GMO: Genmodifisert Organisme